
Automated Analysis
Automated Analysis
Sequencher batch processes your DNA sequence data in a way that is transparent, user definable, and recoverable, and Sequencher never jeopardizes the validity of your scientific conclusions for the sake of automation. Sequencher always gives you the final choice in your sequence editing.
Sequencher always maintains two copies of your data, the edited and the originally imported data. You can undo all or a portion of your edits when you apply the Revert to Experimental Data command to a selection of sequences in your project or to a selection of bases in a sequence.
The Assemble by Name tool allows you to choose a portion of the fragment name to act as a shared identifier, or "assembly handle." Sequencher then makes the selections and names the contigs automatically. Sequencher even supports Regular Expression matching for setting up the unique IDs!
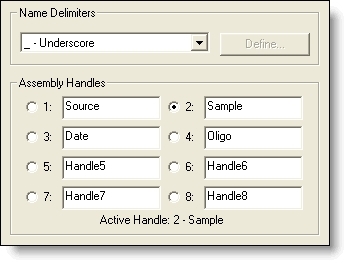
For example, with the click of a button you can convert 90 files, 45 pairs of forward and reverse sequences, into 45 contigs named according to your Patient IDs. A change in your sequence assembly parameters regroups your fragments, so you can assemble the contigs according to Clone ID, Date, Primer, or any other characteristic you record in your sequence names.
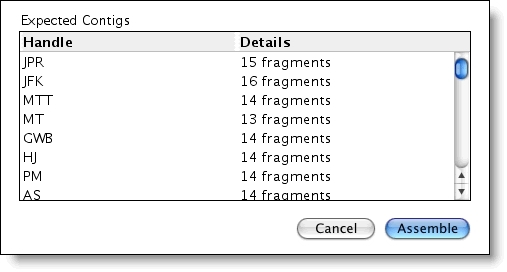
Assemble by Name is particularly helpful if you do a lot of sequencing and if you have numerous samples that are done with a standard set of sequencing primers. Some other applications of Assemble by Name can include:
- Sequencing MHC and mitochondrial genes for identification and population studies
- Analysis of the PCR products of candidate genes.
- Sequencing conserved genes across species for systematics.
- Monitoring of viral genome sequence in conjunction with tracking resistance to antiviral agents or vaccines
The Assemble by Name Tutorial walks you though a typical application of this tool. Additional information about file naming can be found in the Advanced Handle Definition Tutorial.