
Reference-Guided Alignment
Aligners spend a proportion of their time indexing the reference sequence to speed up the overall alignment. Now you can create and keep those indexes (BWA) or databases (GSNAP), making it faster when you want to experiment with different parameters to improve an alignment or splice placement.
The index and database files are cross platform, so you can share them with collaborators on different kinds of computers. When you create a new index or database it will automatically appear in that aligner's list of available reference sequences ready for use at any time. Sequencher remembers where it is, so you don't have to.
The files that come from preprocessing your reference can get large. When you are no longer using one, just delete it with one click using the External Data Browser. You will save disk space and Sequencher can easily recreate it if you need to index the same reference sequence again.
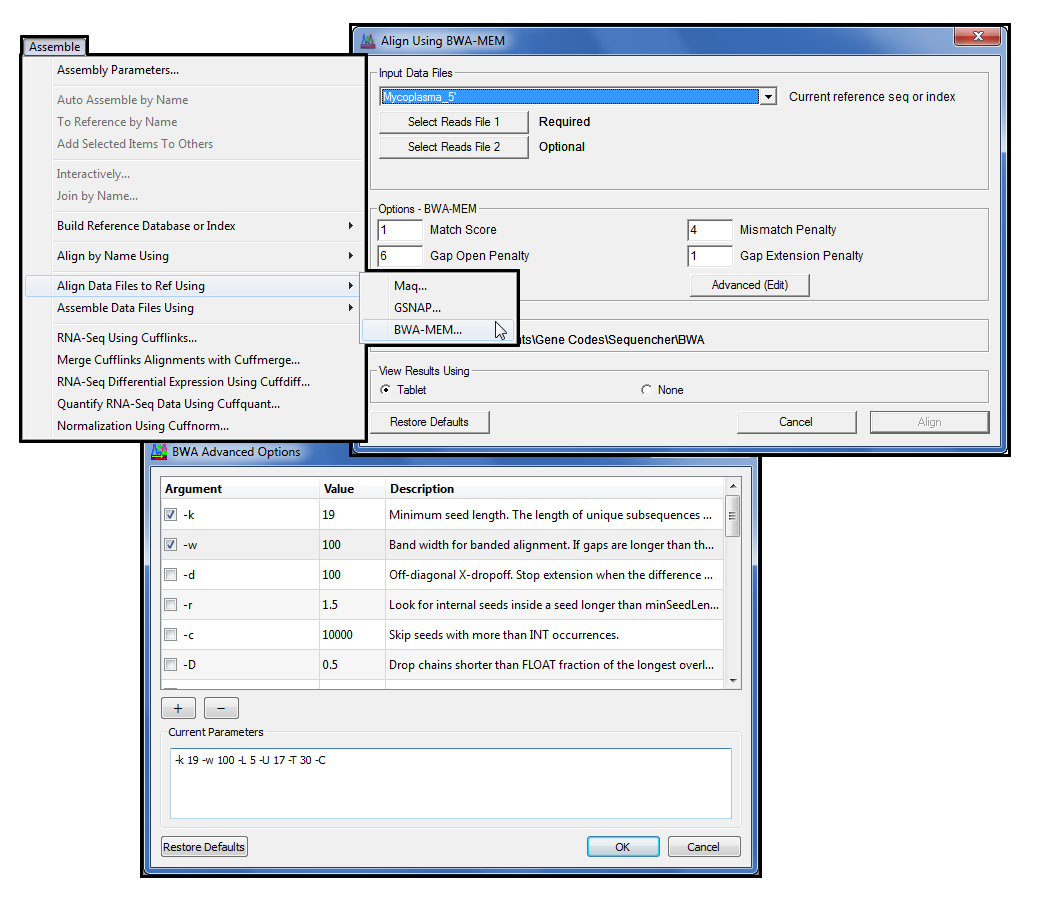
BWA
GSNAP
GSNAP(2) joined the Sequencher family of plugins in version 5.0. GSNAP uses highly efficient methods for reference- based alignment by compressing the reference sequence. This allows GSNAP to be faster and easier while performing Next-Gen sequencing.
GSNAP is designed to preform reference-based alignment of Illumina-Solexa or Sanger standard data that is both paired-ended and unpaired-ended. Length of the sequence is not an issue with GSNAP because it has the ability to align very short to arbitrarily long data lengths with ease.
GSNAP can now work with Multiplex ID (MID) data thereby maximizing efficiency even further. Now you have Sequencher’s user-friendly interface which allows you to harness GSNAP’s power with your MID data. Sequencher automatically divides the data by barcode into separate files, aligns these to the reference using GSNAP, and places the results into separate results folders.
The results of any alignment with GSNAP can be viewed in the popular Tablet browser.

For the power users amongst you who want greater control of GSNAP’s command line functions, press Advanced (Edit) to open the Advanced GSNAP Options dialogue where you may configure specific command line arguments.
Maq
Maq(3)joined the Sequencher family of plugins in version 5.0. The popular Maq algorithm aligns single-end and paired-end Next-Generation data to a reference sequence. Originally designed to align Illumina-Solexa data, it will align any very short read data (63 bases or less). The reference sequence may be in the form of a FastA or GenBank file. Maq uses binary formats to compress the reference and reads files. Best of all, Maq requires very little RAM to run which makes it even easier to perform next-generation sequencing. It also works best with small projects of approximately two million reads, although it is possible to break larger projects down and then merge the results later.
The results of your alignments can be viewed in either Maqview or Tablet.

Originally Maq was written to run on the command line. Sequencher provides a straightforward, easy-to-use interface which protects you from the command line and from having to learn how to use command line arguments.
If you want to learn more about Next-Generation Assembly, check out the Next-Generation Sequence Alignment and Advanced Next-Generation Sequence Alignment Tutorials.
(1)Heng Li Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM 2013 arXiv:1303.3997v2 [q-bio.GN] Please note that this reference is a preprint hosted at arXiv.org.
(2)Thomas D. Wu and Serban Nacu Fast and SNP-tolerant detection of complex variants and splicing in short reads Bioinformatics 2010 26: 873-881
(3)Heng Li, Jue Ruan and Richard Durbin Mapping short DNA sequencing reads and calling variants using mapping quality scores Genome Research 2008 18:1851-1858